PRODUCT MANAGER AT GEICO
Andrew Sungwoo Park

Resistance to HIV due to the CCR5-Δ32 Mutation
CCR5-Δ32 is a deletion mutation in the CCR5 gene. [1] The mutation creates a defective CCR5 co-receptor, and thus prevents human immunodeficiency virus-type 1 (HIV-1) from binding to the receptor sites on cells. The CCR5-Δ32 allele is mainly prevalent in the Northern Europe regions. [2] It is highly likely that selective pressures of many different diseases are responsible for the distribution of the CCR5-Δ32 Mutation. [3]
Contents
1 Entry of HIV
2 Inhibition of HIV through the Mutation
3 Theories on the development of the Mutation
3.1 History of the mutation
3.2 Selective Pressures
4 Treatments utilizing the Mutation
4.1 Stem Cell Transplantation
4.2 Zinc-Finger Nucleases
5 References
Entry of HIV
Once HIV enters the body due to unsafe sex or injection practices, it circulates throughout the body through the bloodstream. HIV has a protective surface studded with a protein called gp-120. The gp-120 of HIV must attach to the CD4 receptor and the CCR5 co-receptor of macrophages in order to enter through endocytosis. [4] The CD4 receptors of macrophages are initial binding sites of HIV. The macrophages also have CCR5 co-receptors that are responsible for penetration and insertion of the RNA strands of HIV [5]. Once HIV has attached and penetrated the macrophage, it releases viral RNA and enzymes into the cytoplasm of the macrophage. The viral enzymes replicate the viral RNA into a double stranded DNA. Then the viral double stranded DNA enters the nucleus and gets integrated into the cell’s DNA. Then the cell undergoes normal transcription and translation. Viral proteins are formed from translation and HIV is created from the host cell and released through exocytosis. This does not affect the host’s cell. [6]
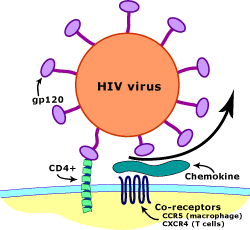
Figure 1. Entry of HIV into Macrophages
As HIV continues to replicate through this cycle, the viral gene undergoes mutation and the mutated gp-120 protein begins to attach to a different co-receptor, CXCR-4, instead of the CCR5 co-receptor. These CXCR-4 co-receptors are found on T cells. The T cells perform transcription and translation of the viral genes as any other cells, but different from the previous exocytosis of HIV, the newly mutated HIV ruptures the membrane of T cells as they leave. [7]
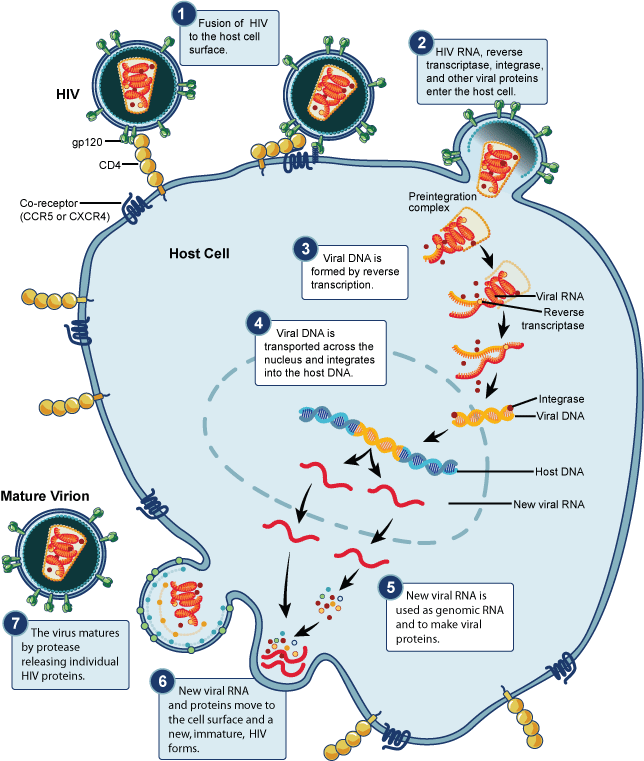
Figure 2. HIV Life Cycle
Inhibition of HIV through the CCR5-Δ32 Mutation
CCR5-Δ32 is a mutation in which a 32-base pair region is deleted from the gene that codes for the CCR5 co-receptor. [8] Individuals with the homozygous or heterozygous mutant alleles have immune signal receptors that are shaped slightly different. [5] HIV has to bind to both the CD4 receptor and the CCR5 co-receptor in order to infect the macrophage, but if the co-receptor is shaped differently, the gp-120 proteins of HIV will not be able to recognize it and not bind to it. If HIV cannot bind to the macrophage, the latter processes of the HIV infection would not occur, thus preventing HIV infection right from its initial entry.
Homozygous carriers of the CCR5-Δ32 mutation are completely resistant to HIV infection because their CCR5 co-receptors are non-functioning, while heterozygous carriers are significantly less susceptible to HIV than those that do not carry this mutation. [5][8] Although both homozygous and heterozygous carriers are immune to HIV, safer sex and injection practices are highly recommended because they can still be infected with strains of HIV that do not enter macrophages through the CCR5 co-receptor. [8] That is due to the fact that HIV is constantly adapting to new environments and evolving.
Theories on the development of the CCR5-Δ32 mutation
History of the mutation
The allele for the Δ32 mutation was dispersed widely at first, spreading over a large geographic area. [2] But over time, the allele frequency increased locally in the northern Europe region due to many selective pressures; and from then on, the mutation remained dormant because it was an unnecessary mutation. [2][3] It was dormant until recently due to the spread of HIV. Because the allele for the Δ32 mutation was so spatially uniform many years ago, there is no geographic restriction on the allele. [2] With time, the Δ32 mutation would definitely spread worldwide.
Selective Pressures
Although the mutation provides people with a resistance to HIV, HIV did not exist in the human population long enough to attribute the mutation to it. That means that there was another selective pressure that is responsible for the distribution of the CCR5-Δ32 mutation. [3]
In the past, many scientists claimed that the Bubonic Plague was the selective pressure that led to the development of the mutation, but that is highly unlikely. The theory is reflective of the intensity of mortality in Europe due to the plague, but the plague did not only affect Europe. [9] In fact, Yersinia pestis, the bacterium that caused the Bubonic plague, spread from Middle East, Africa, and eastern regions. So those regions were probably just as affected by the plague if not more than Europe. This means that the Δ32 mutation could not have been caused by Yersinia pestis, because the high allele frequency for that mutation is located in the northern Europe region and less in the Eastern regions. So many scientists are beginning to think that the Δ32 mutation results from a recurrence of many different diseases such as smallpox and Ebola. [3] Recent studies have shown that the CCR5-Δ32 mutation does not even protect against Yersinia pestis, further proving the fact that the bubonic plague was not the selective pressure for the development of the mutation. [10]

Figure 3. Distribution of CCR5-Δ32 allele frequencies around the world.
Treatments utilizing the CCR5-Δ32 Mutation
Stem Cell Transplantation
In 1995, a patient was diagnosed with HIV. He took medications to decelerate further development of HIV infection. And the same man developed leukemia in 2006 and was in dire need of a bone marrow transplant. His doctor executed a blood stem cell transplant using blood from a donor that had the CCR5-Δ32 mutation. Then the HIV-resistance transferred over to patient and he was cured of HIV. [11]
Although stem cell transplantation is a possible treatment, it is rarely executed successfully. That is due to the fact that the immune cells in the donor may end up attacking the cells in the patient’s body, further deteriorating the patient’s health. [11][12] Also, stem cell transplantation is not a reliable form of treatment because many people have different identifier molecules on their cells. [11] The cells of the donor must closely match up with those of the patients in order for the transplant to be successful. It would be very difficult to find donors that actually match up with the many different patients. [11]
Zinc-Finger Nucleases
An alternative treatment would be to use zinc-finger nucleases (ZFNs) to disrupt the formation of the CCR5 co-receptor. [13] ZFNs permanently disrupt 50% of the CCR5 alleles in the genes so that they would not be able to develop properly. [14] ZFNs are specific enough to only cut out those alleles that code for CCR5 production. Scientists inserted these ZFNs into immune cells and then and inserted those cells into HIV-1 infected mice. The mice showed a decrease in viral replication and increase in T-cells that contain ZFNs. [14] As of now, the splicing of CCR5 allele does not seem to have any detrimental affect when tested on HIV-infected mice. [14] Scientists genetically engineered these ZFNs to mimic the function of the CCR5-Δ32 mutation. [13][14]
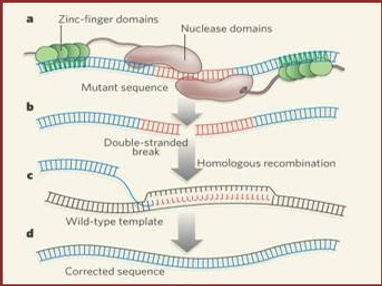
Figure 4. Diagram of Zinc-Finger Nuclease
References
-
Zapico, I., Coto, E., Rodriguez, A., Alvarez, C., Torre, J., & Alvarez, V. (2000). CCR5 (chemokine receptor-5) DNA-polymorphism influences the severity of rheumatoid arthritis.Genes & Immunity, 1(4), 288-289. Retrieved April 24, 2013, from http://www.nature.com/gene/journal/v1/n4/abs/6363673a.html
-
Novembre, J., Galvani, A., & Slatkin, M. (2005). The Geographic Spread of the CCR5 Δ32 HIV-Resistance Allele. PLoS Biology, 3(11). Retrieved April 24, 2013, from http://www.ncbi.nlm.nih.gov/pubmed/16216086
-
Galvani, A., & Slatkin, M. (2003). Evaluating plague and smallpox as historical selective pressures for the CCR5-Δ32 HIV-resistance allele . PNAS, 100(25), 15276-15279. Retrieved April 24, 2013, from http://www.pnas.org/content/100/25/15276 (Links to an external site.)
-
Desormeaux, A., Coutu, M., Medjahed, H., Pacheco, B., Herschhorn, A., Gu, C., et al. (2013). The Highly Conserved Layer-3 Component of the HIV-1 gp120 Inner Domain Is Critical for CD4-Required Conformational Transitions. Journal of Virology, 87(5), 2549-2562. Retrieved April 24, 2013, from http://jvi.asm.org/content/87/5/2549.full (Links to an external site.)
-
Al-Jabri, A. A. (2007). Mechanisms of Host Resistance Against HIV Infection and Progression to AIDS. Sultan Qaboos University Medical Journal, 7(2), 82-96. Retrieved April 24, 2013, from http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3074872/
-
Chan, D. C., & Kim, P. S. (1998). HIV Entry and Its Inhibition. Cell, 93(5), 681-684. Retrieved April 24, 2013, fromhttp://www.sciencedirect.com/science/article/pii/S0092867400814300 (Links to an external site.)
-
Wyatt, R., & Sodroski, J. (1998). The HIV-1 Envelope Glycoproteins: Fusogens, Antigens, and Immunogens. Science, 280(5371), 1884-1888. Retrieved April 24, 2013, fromhttp://www.sciencemag.org/content/280/5371/1884 (Links to an external site.)
-
Marmor, M., Hertzmark, K., Thomas, S. M., Halkitis, P., & Vogler, M. (2006). Resistance to HIV infection. Journal of Urban Health, 83(1), 5-17. Retrieved April 24, 2013, fromhttp://www.ncbi.nlm.nih.gov/pubmed/16736351 (Links to an external site.)
-
Cohn, S., & Weaver, L. (2006). The Black Death and AIDS: CCR5-Δ32 in genetics and history. Q J Med, 99(8), 497-503. Retrieved April 24, 2013, fromhttp://qjmed.oxfordjournals.org/content/99/8/497.full (Links to an external site.)
-
Mecsas, J., Franklin, G., Kuziel, W., Brubaker, R., Falkow, S., & Mosier, D. (2004). Evolutionary genetics: CCR5 mutation and plague protection. Nature, 427(6975). Retrieved April 24, 2013, from http://www.nature.com/nature/journal/v427/n6975/abs/427606a.html
-
Hutter, G., Nowak, D., Blau, I., Hofmann, W., Thiel, E., Mossner, M., et al. (2009). Long-Term Control of HIV by CCR5 Delta32/Delta32 Stem-Cell Transplantation. The New England Journal of Medicine, 360, 692-698. Retrieved April 24, 2013, from http://www.nejm.org/doi/full/10.1056/NEJMoa0802905#t=article (Links to an external site.)
-
Kuo, P., & Stock, P. (2008). Transplantation in the HIV+ Patient. American Journal of Transplantation, 1(1), 13-17. Retrieved April 24, 2013, fromhttp://onlinelibrary.wiley.com/doi/10.1034/j.1600-6143.2001.010104.x/full (Links to an external site.)
-
Holt, N., Wang, J., Cannon, P., Kim, K., Friedman, G., Wang, X., et al. (2010). Human hematopoietic stem/progenitor cells modified by zinc-finger nucleases targeted to CCR5 control HIV-1 in vivo. Nature Biotechnology, 28, 839-847. Retrieved April 24, 2013, from http://www.nature.com/nbt/journal/v28/n8/abs/nbt.1663.html (Links to an external site.)
-
Perez, E., Wang, J., Miller, J., Jouvenot, Y., Kim, K., Liu, O., et al. (2008). Establishment of HIV-1 resistance in CD4+ T cells by genome editing using zinc-finger nucleases.Nature Biotechnology, 26, 808-816. Retrieved April 24, 2013, from http://www.nature.com/nbt/journal/v26/n7/abs/nbt1410.html (Links to an external site.)